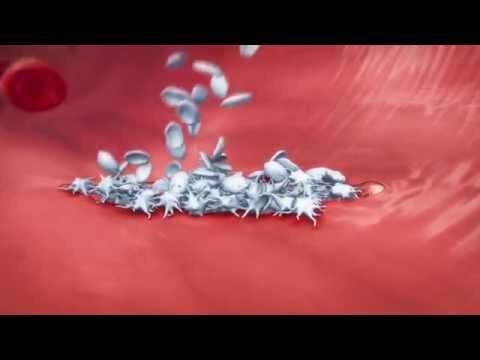
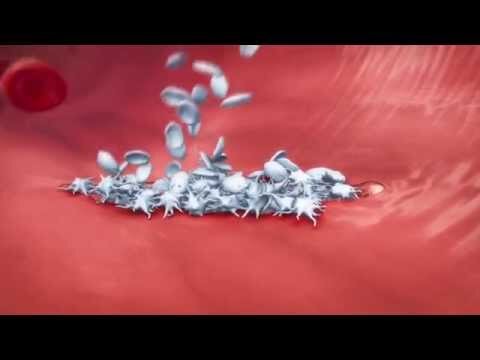
Durable Responses With Adp
Content
The pathologic accumulation and aggregation of α-synuclein (α-syn) underlies Parkinson’s disease . The molecular mechanisms by which pathologic α-syn causes neurodegeneration in PD are not known. Here, we found that pathologic α-syn activates poly(adenosine 5′-diphosphate–ribose) polymerase-1 (PARP-1), and PAR generation accelerates the formation of pathologic α-syn, resulting in cell death via parthanatos. PARP inhibitors or genetic deletion of PARP-1 prevented pathologic α-syn toxicity. In a feed-forward loop, PAR converted pathologic α-syn to a more toxic strain. PAR levels were increased in the cerebrospinal fluid and brains of patients with PD, suggesting that PARP activation plays a role in PD pathogenesis.
These PARP inhibitors prevented α-syn PFF–induced cell death and PARP activation at concentrations as low as 10 nM (fig. S1, C to E). They also reduced α-syn PFF–mediated phosphorylation of α-syn at serine-129 (p–α-syn) (fig. S1, F and G) and α-syn aggregation (fig. S1, H and I), both of which are associated with pathology in α-synucleinopathies .
How Can Parents And Teachers Help?
However, the main role of ADP-ribosylated ubiquitin generated by SidE proteins is to serve as an activated intermediate for non-canonical serine ubiquitylation of host proteins that is important for supporting bacterial infection. Unconjugated polyubiquitin chains are present in cells , so it is conceivable that the glycine 76 carboxyl group at the C-terminus of the polyubiquitin chain can be modified by Dtx3L/Parp9. However, to the best of our knowledge, handling of free polyubiquitin chains by E1 and E2 has not been observed, and thus, ADP-ribosylation of free polyubiquitin chains by Dtx3L/Parp9 remains highly speculative. On the other hand, SidE proteins can modify existing polyubiquitin chains. Incubation of lysine 48- or methionine 1-linked ubiquitin tetramers with SdeC led to ADP-ribosylation of the incorporated ubiquitin .
Mice treated with ABT-888 exhibited significantly less loss of DA neurons after an intrastriatal injection of α-syn PFF compared to mice on the control diet (Fig. 3, B and C). ABT-888 also reduced the formation of α-syn PFF–induced increase in PAR levels (fig. S5A). Tyrosine hydroxylase and DA transporter levels were also reduced in WT mice in response to α-syn PFF, whereas the reduction in TH and DAT levels was prevented in PARP-1 KO or ABT-888–treated WT mice (fig. S5, A to C). The loss of DA neurons was accompanied by a reduction in striatal DA and its metabolites in WT mice, but not PARP-1 KO or ABT-888–treated mice (Fig. 3D and fig. S5, F to H).
- α-Synuclein PFF was found to primarily kill neurons via parthanatos, because necroptosis and autophagy inhibition had no effect on α-synuclein PFF neurotoxicity and there was only modest protection by caspase inhibition.
- We investigated the cellular cell death pathways that contribute to and drive α-synuclein PFF–mediated neuronal cell death.
- Recombinant α-synuclein preformed fibrils , which are similar in structure to those found in PD, were used to model pathologic α-synuclein both in vitro and in vivo.
- Pathologic α-synuclein was found to activate nitric oxide synthase , causing DNA damage and poly(adenosine 5′-diphosphate–ribose) polymerase-1 (PARP-1) activation, leading to cell death via parthanatos.
PAR generated by PARP-1 activation also binds to α-synuclein, accelerating its fibrillization and converting pathologic α-synuclein to a more misfolded compact strain with 25-fold enhanced toxicity. PAR-modified α-synuclein PFF–injected mice showed accelerated disease progression and phenotype compared to α-synuclein PFF–injected mice. Moreover, PAR levels were increased in the cerebrospinal fluid in two independent patient cohorts and brains of PD patients, providing evidence that parthanatos may contribute to the pathogenesis of PD. The main outcome of this project was to determine that disruption of poly(ADP-ribosyl)ation mechanisms alters responses of Arabidopsis thaliana to biotic stress. Poly(ADP-ribosyl)ation is a post-translational protein modification in which ADP-ribose units derived from NAD+ are attached to proteins by poly(ADP-ribose) polymerase enzymes. In animals, poly(ADP-ribosyl)ation is associated with DNA damage responses and programmed cell death. We initially hypothesized a role for poly(ADP-ribosyl)ation in plant defense responses when we detected the up-regulated expression of Arabidopsis ADP-ribosylation-related genes upon infection with avirulent Pseudomonas syringae pv.
Activation of parthanatos is the primary driver of pathologic α-synuclein neurodegeneration. Inhibition of PARP and depletion of PARP-1 substantially reduces the pathology induced by the transmission of pathologic α-synuclein. In a feed-forward loop, PAR converted pathologic α-synuclein to a more toxic strain and accelerated neurotoxicity both in vitro and in vivo. Consistent with the notion that PARP-1 activation plays a role in PD pathogenesis, PAR levels were increased in the CSF and brains of PD patients. Thus, strategies aimed at inhibiting PARP-1 activation could hold promise as a disease-modifying therapy to prevent the loss of DA neurons in PD and related α-synucleinopathies.
As previously described , a single intrastriatal injection of α-syn PFF leads to an approximate 50% loss of DA neurons 6 months after the injection in wild-type mice (Fig. 3, B and C). In contrast, a single intrastriatal injection of α-syn PFF into PARP-1 KO mice failed to induce DA cell loss (Fig. 3, B and C). WT mice were also fed a diet containing the PARP inhibitor ABT-888 (125 mg/kg) and compared with mice given a control diet (Fig. 3, B and C).
Experiments with gallotannin were also conducted to block poly(ADP-ribose) glycohydrolase activity. To determine whether α-syn PFF induces the activation of PARP, levels of PAR were measured using a highly sensitive and specific PAR monoclonal antibody after administration of α-syn PFF to primary mouse cortical neurons (Fig. 1). α-Syn PFF (1 μg/ml) induced PARP activation peaks between 3 and 7 days and remained elevated for up to 14 days (Fig. 1A).
What Factors, If Any, Might Affect The Lab Results? In Particular, Does Your Patient Take Any Medications
As previously described , injection of intrastriatal α-syn PFF leads to α-syn pathology in DA neurons of WT mice (figs. S5, A, D, and E, and S6, A to D). α-Syn pathology was markedly reduced in PARP-1 KO mice and ABT-888–treated WT mice consistent with the absence and reduction of neurodegeneration, respectively. Intrastriatal injection of α-syn PFF caused deficits on the pole test , a sensitive behavioral measurement of dopaminergic function, in WT mice, whereas there were no deficits in PARP-1 KO and ABT-888–treated WT mice (Fig. 3E and fig. S6E). Both forelimb plus hindlimb and forelimb grip strength were also reduced in WT mice after α-syn PFF injection, but not in PARP-1 KO or ABT-888–treated WT mice (Fig. 3F and fig. S6F). Thus, the striatal α-syn PFF–induced loss of DA neurons is dependent on PARP-1. Ubiquitylation is a post-translational modification that regulates a wide range of cellular pathways including protein degradation, autophagy, mitophagy, cell signaling, DNA damage response, and protein trafficking.
Treatment of cortical neurons with the broad-spectrum caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK) (Z-VAD) partially reduced α-syn PFF toxicity. The necroptosis inhibitor necrostatin-1 (Nec-1) and the autophagy inhibitor 3-methyladenine (3-MA) had no effect, whereas the PARP inhibitor ABT-888 prevented α-syn PFF toxicity (fig. S2, G to M). Because PARP inhibition and knockout of PARP-1 reduced the accumulation of pathologic α-syn as indicated by a reduction of p–α-syn immunostaining, we assessed cell-to-cell transmission of α-syn . Knockout of PARP-1 or PARP inhibition did not show a difference in the levels of α-syn–biotin PFF in endosomal-enriched fractions (fig. S3, A to D) , indicating that PARP-1 did not affect the uptake of α-syn PFF. However, knockout of PARP-1 reduced the cell-to-cell transmission of pathologic α-syn by inhibiting propagation of α-syn PFF into recipient cells (fig. S3, E to G).
α-Synuclein PFF was found to primarily kill neurons via parthanatos, because necroptosis and autophagy inhibition had no effect on α-synuclein PFF neurotoxicity and there was only modest protection by caspase inhibition. Neuron-to-neuron transmission of pathologic α-synuclein and accompanying pathology and neurotoxicity in primary neuronal cultures were completely attenuated by clinically available PARP inhibitors or by deletion of PARP-1. α-Synuclein PFF–induced loss of DA neurons and biochemical and behavioral deficits in vivo were significantly prevented by PARP inhibition or lack of PARP-1.
Deletion or knockout of PARP-1 prevented α-syn PFF–mediated PARP activation and cell death (Fig. 1, E to H, and fig. S2, A and B). Knockout of PARP-1 also reduced p–α-syn immunostaining and α-syn aggregation (fig. S2, C to F).
tomato DC3000 avrRpt2+, and pathogen-dependent changes in the poly(ADP-ribosyl)ation of discrete proteins were also observed. We concluded that poly(ADP-ribosyl)ation is a functional component in plant responses to biotic stress. The 2010 Ph.D. thesis of Amy Briggs (in preparation for peer-review publication) reports an extensive gene expression profiling experiment using Nimblegen whole-genome Arabidopsis arrays. Responses to flg22 in the presence/absence of 3AB were monitored, as were responses of wild-type and parg1 mutants to elf18.
Human Csf Samples And Par Elisa
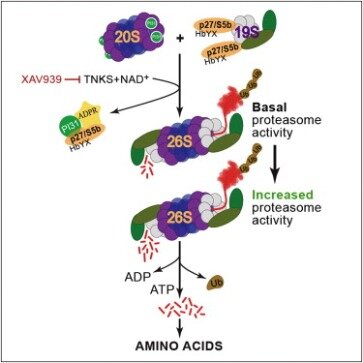
Moreover, assessment of PAR levels in the CSF could serve as a theranostic biomarker for disease-modifying therapies in these disorders. ADP-ribosylation of ubiquitin is an exciting new addition to the possible post-translational modifications for ubiquitin. To date, SidE proteins and the Dtx3L/Parp9 complex are the only proteins known to generate ADP-ribosylated ubiquitin. These two cases of ADP-ribosylation of ubiquitin represent distinct mechanisms by which ubiquitin conjugation activity is regulated . Dtx3L/Parp9-mediated ADP-ribosylation of the C-terminal glycine 76 of ubiquitin effectively prevents the modified ubiquitin from being utilized in ubiquitylation processes until the blocking ADP-ribose is removed.
If our model is correct, then ISG transcription should be increased because the negative regulatory mechanism on Dtx3L E3 activity is relieved by inactivation of Parp9 catalytic activity. Similar to ubiquitin ADP-ribosylated at the C-terminal glycine 76, ubiquitin ADP-ribosylated on arginine 42 and its phosphoribosylated derivative are unable to be utilized in E1 and E2 processing, thereby disrupting host ubiquitylation processes .
Mutant parg1 seedlings exhibit an exaggerated seedling growth inhibition and pigment accumulation in response to elf18, and are hypersensitive to the DNA damaging agent mitomycin C. Both parg1 and parg2 knockout plants show accelerated onset of disease symptoms when infected with Botrytis cinerea. Cellular levels of ADP-ribose polymer increase after infection with avirulent Pseudomonas syringae pv.
The elevation of PAR was accompanied by neuron death as assessed by propidium iodide staining (Fig. 1, B and C). Treatment of cortical neurons with 1 μM of the PARP inhibitors [ABT-888 , AG , or BMN 673 ] prevented the α-syn PFF–mediated PARP activation and cell death (Fig. 1, B to D). Complete inhibition was observed at higher concentration of these inhibitors (fig. S1, A and B).
Arabidopsis NUDT7 mutants supported reduced growth of virulent P. syringae as also reported by others, but we also found a loss-of-hypersensitive-response phenotype. Inhibitors of poly(ADP-ribose) polymerase disrupted FLS2-mediated basal defense responses such as callose deposition. EIN2 , IXR1 and IXR2 mutants impacted the FLS2-mediated responses that occur during PARP inhibition, while no impacts were observed for NPR1, PAD4 or NDR1 mutants. Further work defined aspects of poly(ADP-ribosyl)ation during plant defense responses in soybean and tomato . The role of poly(ADP-ribosyl)ation in plant defenses was more extensively investigated using Arabidopsis thaliana.
Thus, strategies aimed at inhibiting PARP-1 activation could hold promise as a disease-modifying therapy to prevent the loss of dopamine neurons in PD. While both the Dtx3L/Parp9 complex and the SidE proteins can mediate the formation of ADP-ribosylated ubiquitin, clear distinctions are evident between the two cases. The ADP-ribosylation of ubiquitin by Parp9 requires that ubiquitin undergo E1 and E2 processing as well as the presence of the E3 ubiquitin ligase Dtx3L. In contrast, the mART domain of SidE proteins is sufficient to generate ADP-ribosylated ubiquitin in the absence of ubiquitin processing by E1 and E2. The roles of the generated ADP-ribosylated ubiquitin are different between Dtx3L/Parp9 and SidE proteins. For Dtx3L/Parp9, ADP-ribosylation of ubiquitin takes place to short-circuit the normal ubiquitylation process by Dtx3L, thereby acting in a negative regulatory manner.
This post-translational modification is characterized by covalent attachment of ubiquitin to lysine residues on target proteins by E3 ubiquitin ligases. These enzymes can catalyze both mono- and polyubiquitylation of target substrates.
Further investigation showed that ADP-ribosylation and phosphoribosylation of lysine 63-, lysine 48-, lysine 11-, or methionine 1-linked diubiquitin chains could be catalyzed by SdeA . No preference for modification of either ubiquitin within the diubiquitin was observed. Because synthetic α-syn PFF killed primary cortical neurons via parthanatos, we sought to determine whether parthanatos plays a role in the loss of dopamine neurons after the intrastriatal injection of α-syn PFF (Fig. 3) . A single intrastriatal injection of α-syn PFF (5 μg) induced PARP activation and increased PAR levels (Fig. 3A). Intrastriatal injection of α-syn PFF into PARP-1 KO mice failed to increase PAR levels (Fig. 3A).
It is possible that the control of Dtx3L-mediated ubiquitylation by Parp9-mediated ADP-ribosylation of ubiquitin operates in the context of active IFN signaling and viral infection as well. Depletion of NAD+ has been observed in cells treated with IFNγ , and this could further augment Dtx3L E3 output and downstream control of viral infection. Decreasing levels of NAD+ would lead to less Parp9-mediated ADP-ribosylation of ubiquitin; hence, unmodified ubiquitin is available for Dtx3L-mediated ubiquitylation of histone H2BJ which results in increased ISG expression. It would be interesting to test this hypothesis through co-expression of catalytically inactive Parp9 mutant and Dtx3L and examine how transcription of ISG is impacted.
Dtx3l E3 Output Is Regulated By Adp
Recombinant α-synuclein preformed fibrils , which are similar in structure to those found in PD, were used to model pathologic α-synuclein both in vitro and in vivo. We investigated the cellular cell death pathways that contribute to and drive α-synuclein PFF–mediated neuronal cell death. Pathologic α-synuclein was found to activate nitric oxide synthase , causing DNA damage and poly(adenosine 5′-diphosphate–ribose) polymerase-1 (PARP-1) activation, leading to cell death via parthanatos.
Because of the presence of multiple ubiquitylation acceptor sites on ubiquitin, polyubiquitin chains differing by linkage type and branching patterns can be generated. Post-translational modifications on ubiquitin including glutamine deamidation, lysine SUMOylation, lysine acetylation, and serine, threonine, and tyrosine phosphorylation add to the range of ubiquitin structures that can be synthesized in cells. Recently, ADP-ribosylation was discovered as a new post-translational modification on ubiquitin in two different biological contexts. The bacterial SidE proteins ADP-ribosylate ubiquitin to activate it for a unique mode of ubiquitylation. The human Dtx3L /Parp9 (ADP-ribosyltransferase) complex ADP-ribosylates ubiquitin which inhibits conjugation. We identified PARP-1 activation and the generation of PAR as a key mediator of pathologic α-synuclein toxicity and transmission.
